
Нобелевскую премию по химии присудили за решение «проблемы белка». Ученые бились над ней полвека, а лауреатами в итоге стали программисты Google Но в выигрыше все: открытие ускорит разработку лекарств
Нобелевскую премию по химии в 2024 году присудили трем исследователям: половину — Дэвиду Бейкеру из Университета Вашингтона за «компьютерный дизайн белков»; вторую половину — двум сотрудникам DeepMind, Демису Хассабису и Джону Джамперу за «предсказание трехмерной структуры белка». Обе части награды относятся к одной и той же теме, известной в науках о жизни как «проблема белка». В рамках ее решения ученые пытались понять, как взаимосвязаны химический состав этих молекул и их трехмерная структура.
Первым важный шаг в нужном направлении сделал Бейкер. Еще в начале 2000-х он решил «обратную версию» проблемы, то есть научился предсказывать, какая аминокислотная последовательность потребуется для того, чтобы получить белок с некоей новой, не существующей в природе трехмерной структурой. Проблему же белка в ее исходной версии, то есть задачу предсказания структуры на основе последовательности, удалось решить Хассабису, Джамперу и всей команде DeepMind несколько лет назад. В конце 2020 года подразделение Google обнародовало результаты тестирования программы AlphaFold2, сделанной на основе нейросетей. Она оказалась способна предсказывать структуру белков с невиданной ранее точностью почти в 90% — зачастую лучше, чем обычные химические методы. Спустя несколько месяцев в DeepMind предсказали структуры сотен тысяч белков человека и еще 20 модельных организмов. А в последующие годы AlphaFold2 стала общим стандартом и уже широко используется в поиске лекарств.
О том, какую революцию произвела в структурной биологии AlphaFold2, как она устроена, в чем состоит ее влияние на простых ученых и причем здесь японская игра го, «Медуза» писала по горячим следам открытия в 2020 году. Этот рассказ остается полностью релевантным. Мы публикуем его без изменений, но с незначительными пояснениями.
Этот текст впервые опубликован 13 декабря 2020 года.
Жизнь, по Энгельсу, — это форма существования белковых тел. Поэтому понимать, как работают белки, очень важно. Ученые научились хорошо и быстро читать геномы, в которых закодирована информация о белках. Устанавливать аминокислотные последовательности белков просто, но понять, какую форму они принимают в клетке и как работают, мы до сих пор не умеем — это и есть «проблема белка». Ученые точно знают, что, прочитав геном и узнав последовательность белка, можно — теоретически — предсказать его структуру, но сделать это до сих пор не получается. Сейчас ученым приходится не предсказывать, а напрямую определять структуру, тратя на это многие годы. Это сложно, но важно, потому что только так можно понять, как именно лекарство действует на свою мишень, которая почти всегда представляет из себя белок.
Теперь все поменялось: в недрах Google появилась программа, которая может по аминокислотной последовательности белка построить его трехмерную структуру. Это работает очень точно и почти для любой аминокислотной последовательности. В практическом плане это означает, что будь такой алгоритм доступен раньше, подбор лекарства против коронавируса мог бы начаться в ту же минуту, как был прочитан его геном. Этот, самый первый этап разработки лекарств, теперь может быть сильно ускорен. Но повлияет ли это на стоимость и скорость разработки лекарств в целом, пока не ясно.
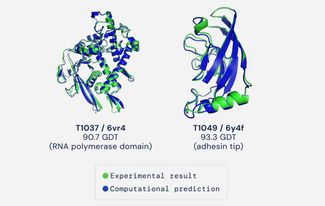
«Сначала мы пытались определить структуру нашего белка с помощью традиционного рентгеноструктурного анализа» — рассказывает Мария Соколова, старший преподаватель Сколтеха, чья работа стала яркой иллюстрацией новых возможностей компьютерного подхода к предсказанию белковых структур. Иллюстрацией в буквальном смысле — именно структура, найденная Марией с соавторами в ходе исследования одной из вирусных полимераз, появилась на страницах всех мировых СМИ, которые написали о новом успехе Google. Белок, о котором идет речь — это РНК-полимераза бактериального вируса phi14:2, молекулярная машина, которую вирус использует для переноса своей генетической информации с носителя на носитель — с ДНК на РНК. Подобные ферменты часто становятся мишенями для новых лекарств, но для команды Deepmind это было не важно — структура выступила в роли золотого стандарта, с которым сравнивалось предсказание, сделанное программой AlphaFold2. Если взглянуть на изображение, где две структуры наложены друг на друга, то заметить хоть какие-то отличия между ними невооруженным глазом почти невозможно. Но чего на изображении не видно совсем — так это колоссальной разницы в усилиях, которые потребовалось на их получение.
«Для рентгеноструктурного анализа нужно вырастить из белка кристаллы, поместить один из них под пучок рентгеновского излучения (на синхротроне) и собрать данные дифракции пучка, обработав которые, можно определить структуру. Процесс кристаллизации белка — сложный и непредсказуемый. Миллионы индивидуальных молекул белка должны уложиться в идеальный трехмерный массив. Малейшее отклонение, например, из-за того, что у белка есть подвижные части, — и кристалл уже не даст хорошую дифракцию. Какие-то белки легко кристаллизуются, а какие-то — нет, и ученые десятилетиями не могут определить их структуру» — объясняет Мария.
За год стажировки в лаборатории Петра Леймана в Университете Техаса, где Мария также работала над структурой полимеразы вируса AR9, ей пришлось вырастить тысячи кристаллов в самых разных условиях, проанализировать дифракцию нескольких сотен из них на синхротроне, но даже всех этих данных было недостаточно для того, чтобы построить атомную модель белка. Тогда исследователям пришлось переключиться на совершенно другой метод, который не требует получения капризных кристаллов, и который в последние годы стал особенно популярен среди специалистов — криоэлектронную микроскопию. Этот подход заключается в анализе снимков отдельных белковых молекул, замороженных на подложке внутри микроскопа. Плоские фото россыпи отдельных молекул затем собираются в объемную структуру с помощью сложного алгоритма — задача напоминает построение трехмерной модели Эйфелевой башни из тысяч разрозненных фотографий, сделанных вокруг нее туристами. С тем, конечно, отличием, что Эйфелева башня в Париже одна, а белковых молекул на подложке — много, но они повернуты в разных ракурсах.
«Там [с криоэлектронной микроскопией] тоже были свои подводные камни и проблемы, но мы их все-таки сумели решить, — лаконично резюмирует Мария. — В итоге мы собрали огромное количество данных и построили на их основе атомную модель нашего фермента практически целиком, за исключением некоторых неупорядоченных фрагментов. Вся эта работа заняла у нас более трех лет».
Когда структура все-таки была получена, в конце зимы 2020 года, Петр Лейман, руководитель лаборатории, где Мария билась с решением проблемы белка на своем участке фронта, отправил последовательности нескольких изучаемых белков (в том числе phi14:2 и AR9 полимераз) на конкурс CASP14. Это традиционное соревнование алгоритмов, занимающихся предсказанием структуры по аминокислотной последовательности. Именно в нем в этом году победила созданная командой DeepMind программа AlphaFold2, оставив далеко позади всех конкурентов.
«Когда наши последовательности были включены в соревнование, мы совсем не ожидали, что фрагменты, структуру которых мы пока не сумели построить даже имея какие-то экспериментальные данные, могут быть пересказаны компьютерным алгоритмом „с нуля“» — рассказывает Мария. — «Но, к нашему абсолютному удивлению, произошло именно это — компьютер предсказал структуру всех белков абсолютно правильно. Включая те домены и белки, которые были непохожи ни на что известное науке до сих пор!».
В чем ваша проблема?
Сделанный Google прорыв в предсказании белковых структур на практике означает то, что теперь трехлетнюю работу ученых-исследователей можно упаковать в несколько дней вычислений на не самом мощном компьютере. Это, безусловно, впечатляет. Но прежде, чем делать прогнозы о том, как AlphaFold2 повлияет на биологию и медицину, хорошо бы сказать пару слов о самой проблеме белка и о природе ее сложности.
Сложность эта происходит из огромного, фантастического числа возможных вариантов трехмерной укладки одной белковой цепи, среди которых алгоритму требуется найти правильное решение. С точки зрения вычислений здесь ситуация в точности напоминает ту, которая сложилась с игрой го. До того, как в 2016 году предыдущее детище команды DeepMind, программа AlphaGo, расправилась с одним из сильнейших игроков на планете Ли Седолем, большинство экспертов в теории игр сходилось в том, что эта «проблема го» попросту нерешаема. В отличие от шахмат, в го число возможных комбинаций камней на доске настолько велико, что перебором его просчитать гарантированно невозможно — оно превышает число атомов в наблюдаемой вселенной. Однако для того, чтобы победить сильнейших в мире игроков, этого делать не потребовалось. Вместо перебора, AlphaGo, используя мощь архитектуры сверточных нейросетей (тех самых, что применяются в распознавании изображений и их генерации), научилась анализировать локальную ситуацию на каждом участке доски, не просчитывая до конца варианты развития событий — примерно так, как это делают и обычные игроки. Для этого ей, конечно, понадобилось пройти обучение на записях игр живых людей. Не сплошной перебор, а именно такой анализ («при таком ходе мы, скорее, выиграем, а не проиграем») позволил программе с разгромным счетом победить Ли Седоля, который впоследствии и вовсе бросил профессиональный спорт.
Расправившись с го, команда DeepMind, возглавляемая Демисом Хассабисом, переключилась на проблему белка. Даже по сравнению с го эта задача формально существенно сложнее — просто из-за числа вариантов решения. Белки состоят из цепочек аминокислот, и размер такой цепочки обычно колеблется от нескольких десятков до нескольких сотен звеньев. «Свернуться в клубок» такая цепь может огромным количеством способов. В 1969 году молекулярный биолог Сайрус Левинталь оценил, что для типичного белка количество возможных вариантов пространственной укладки составляет 10 в трехсотой степени — для сравнения, это на 129 порядков больше, чем число комбинаций в го. Ожидать постепенного перебора всех этих вариантов можно было бы миллиарды лет — но в реальной клетке (да и просто в пробирке), аминокислотная цепочка принимает нужную форму всего за доли секунды. Такое яркое несоответствие реальной и ожидаемой скорости сворачивания белка получило название парадокса Левинталя. То, что природа решает эту проблему гораздо быстрее, чем кажется возможным, означает, что здесь есть какие-то скрытые фундаментальные законы — их-то и пытались вывести ученые в течение последующих 50 лет.
С успехами в поиске простых и понятных законов укладки белковой цепи до сих пор было совсем плохо. Но поскольку трехмерные структуры для понимания того, как работают молекулярные машины все-таки нужны, ученые использовали для их определения не предсказание, а прямые физические методы — прежде всего рентгеноструктурный анализ, с которого начала команда Марии Соколовой, ядерно-магнитный резонанс и, в последние годы, криоэлектронную микроскопию. Эти методы требуют очень много времени и сил, но, кроме того, с их помощью можно определить структуру далеко не каждого белка — особенно трудно поддаются те белки, которые живут на поверхности клеток, плавая в клеточной мембране. Их жирные, отталкивающие воду участки не дают им нормально существовать в растворе и мешают собираться в кристаллы. Однако те же мембранные белки составляют очень важную часть лекарственных мишеней — рецепторы гормонов, нейромедиаторов, даже «входные ворота» нового коронавируса ACE2 — все это мембранные белки. Таким образом, даже несмотря на постоянный и значительный прогресс в развитии физических методов, ученым по-прежнему очень нужен вычислительный алгоритм, который бы мог предсказывать правильную трехмерную структуру белков только на основе последовательности их аминокислотной цепи. Решение этой проблемы, назвав ее «основополагающей проблемой биологии», и пытались приблизить ученые, положившие начало конкурсу алгоритмов CASP, на котором в этом году победила AlphaFold.
У конкурса довольно длинная история. В 1994 году его учредили Джон Молт из Университета Мериленда и Кшиштоф Фиделис из Калифорнийского университета в Девисе. По правилам соревнования, разные команды программистов пытаются предсказать с наибольшей возможной точностью впервые открытые структуры белков, — те, которые уже определены традиционными физическими методами, но еще не опубликованы в научной литературе. Получился своего рода конкурс-угадайка: организаторы знают от независимых исследователей правильный ответ на вопрос о структуре нескольких белков, участники должны их угадать — а побеждает тот, кто сделает это как можно точнее.
Для того, чтобы понять, насколько близко участники приблизились к ответу, Молт ввел понятие «теста глобального расстояния» (Global Distance Test, GDT), который отражает, насколько близко координаты ключевых атомов углерода в полученной структуре совпадают с теми координатами, которые установлены в эксперименте. Поскольку речь о доле атомов, угаданных достаточно точно (с определенной отсечкой), то максимальное значение GDT составляет 100 процентов. Впрочем, уже изначально, учитывая возможность наличия ошибок в самих физических методах, Молт и Фиделис посчитали, что определением полного успеха для вычислительного алгоритма можно считать 90%-е попадание, то есть GDT 90.
Как в ее решении продвинулись ученые из DeepMind
Как и AlphaGo, AlphaFold2 — это программа, в сердце которой обученная сверточная нейросеть довольно сложной архитектуры. Детали ее устройства до конца не ясны, поскольку подробная научная публикация по результатам конкурса еще не вышла — пока всем приходится ориентироваться на довольно скупой на технические подробности пресс-релиз. Но из него, а также из описания первой версии AlphaFold, которое было опубликовано в Nature еще в 2018 году, можно примерно представить, как она работает.
Сеть использует информацию о 170 тысячах структур, доступных в мировой базе белков PDB, но кроме того, — и это, по-видимому, было крайне важно для победы — учитывает не только геометрию структур, но и эволюционное сходство между похожими белками из разных организмов. Идея, которая стоит за таким подходом, довольно проста: поскольку задача в том, чтобы предсказать трехмерную структуру, а в ней удаленные друг от друга (по последовательности) аминокислоты обычно соприкасаются между собой, то для решения проблемы белка очень важно прежде всего найти такие дальние взаимодействия. Если их установить, ученые смогут сделать что-то вроде реконструкции костюма инопланетянина по выкройке: даже если вы не знаете, где на плоском рисунке обозначен рукав, а где штанина — вы все равно сможете найти соответствие между удаленными друг от друга пуговицей и петлей, и соединив их, восстановить хотя бы локальную геометрию. Сложность в том, чтобы найти эти пары. Именно здесь помогает анализ родственных белков: имея сходные последовательности из разных организмов, где одни аминокислоты в ходе мутагенеза постепенно заменились другими, можно попытаться вычислить такие взаимодействующие пары. В ходе эволюции они будут меняться согласованно друг с другом — как бы ни менялась мода, поменять местами пуговицу и петлю (и сохранить взаимодействие) существенно легче, чем пристегнуть штанину к воротнику (и все сломать).
Улучшенная версия алгоритма, AlphaFold2, учитывает в своей работе именно эту особенность белковой эволюции. И если в 2018 году на 13-й конференции CASP AlphaFold лишь ненамного превзошла конкурентов, то в этом году программа достигла золотого стандарта и определила предлагаемые структуры со средним результатом GDT 92,4. В категории «свободного моделирования» без каких-либо вводных, то есть зная только аминокислотную последовательность, программа набрала чуть меньше очков, хотя тоже немало — 87% атомов находились на своих местах. Разработчики оценили разрешение полученных компьютером структур в 1.6 ангстрем (1 ангстрем это 10 в -10 метра, что примерно равно расстоянию между атомами в большинстве кристаллов). И если 87 очков кажется слишком маленьким значением (меньше 90, о которых мечтали) то надо напомнить, что, вообще говоря, это даже лучше, чем большинство структур, полученных при помощи традиционного рентгена. По сути, DeepMind удалось создать программу, которая (наконец-то) может заменить все трудоемкие физические методы определения структуры недолгими расчетами
«Те фрагменты, которые нам удалось определить самим, были предсказаны AlphaFold2 очень точно, — продолжает Мария Соколова. — Но самое главное, те части, для которых мы еще даже не закончили создание атомной модели, тоже были предсказаны, и отлично вписывались в наши экспериментальные данные, карту электронной плотности. Коллеги были в шоке, увидев это. Представьте, вы сидите месяцами, а то и годами, пытаясь собрать пазл из экспериментальных данных, полученных на очень дорогом оборудовании, и построить модель белка — и тут вдруг открываете результат нескольких дней работы компьютера, который сравним или превосходит то, что вы делали всё это время…»
А есть ли от этого практическая польза?
В том, что новая версия AlphaFold представляет собой безусловный алгоритмический прорыв и еще одну победу нейросетей, согласны почти все. Но главный вопрос, конечно, заключается не в том, насколько новый алгоритм лучше старых — а в том, насколько появление таких программ может ускорить фундаментальную науку и процесс разработки лекарств.
В Google свой успех подают именно как важнейший этап на пути к этой цели. Однако тут взгляды экспертов уже сильно расходятся. Мария Соколова считает, что AlphaFold — это настоящий прорыв для ученых-структурщиков, и не только. «Фундаментальная наука безусловно шагнет вперед. Например, теперь можно будет предсказать структуры белков с ранее неизвестными функциями и обнаружить прежде неизвестные эволюционные связи между ними, которые не видно по аминокислотной последовательности этих белков. Так, например, только определив структуру полимеразы бактериального вируса, мы поняли, что она родственна другой полимеразе из высших организмов, которая там отвечает за процесс РНК-интерференции (которой у бактерий нет). Это стало открытием достойным публикации в самом престижном научном журнале, — Nature». Коллега Соколовой, Петр Лейман даже высказал мнение, что искусственный интеллект произведет в биологии такую же революцию, какую произвела в физике теория относительности.
Однако другие специалисты настроены менее оптимистично. «Кажется, что первоочередная польза [от AlphaFold2] будет для структурщиков-экспериментаторов. Во всех их методах нужна некоторая стартовая модель, которая уже будет улучшаться за счет наложенных экспериментальных данных, — комментирует Артур Залевский, преподаватель факультета биоинженерии и биоинформатики МГУ. — В самостоятельной пользе моделей, полученных этим путем я очень сильно сомневаюсь».
Дело, по словам Залевского, не в самих алгоритмах, а в том, что база данных о белковых структурах, на которой они обучаются, содержит (вполне ожидаемо), примеры только тех из них, для которых довольно просто получить кристаллы. Кроме того, действительно хороших структур, с разрешением в один ангстрем, там очень мало, менее тысячи, а качество остальных ощутимо хуже. Все это делает предсказания AlphaFold2 хорошими лишь по сравнению с теми в среднем плохими структурами, которые ученые смогли определить к настоящему моменту.
По мнению Залевского, для того, чтобы искусственный интеллект действительно совершил в структурной биологии революцию, нужно преодолеть еще длинный путь. Нужны новые, качественные структуры, причем полученные в условиях, максимально приближенных к тем, в которых находятся живые клетки — а не в тех, в которых хорошо растут кристаллы. «Машинное обучение в биологии хронически страдает от недостатка отрицательных контролей — в существующих базах мы видим только то, что можем увидеть, что удалось кристаллизовать/сделать ЯМР. То, что плохо синтезируется, не сворачивается или сворачивается не так, как нужно для получения кристалла, — всего этого мы не видим. А это ценнейшая информация для машинного обучения.»
Однако, даже если не учитывать эти тонкости и допустить, что на самом деле предсказываемые AlphaFold2 структуры на самом деле очень точны, остается вопрос о том, насколько сильно они смогут изменить не теорию, а практику разработки лекарств. Является ли получение структуры белка тем самым узким местом, из-за которого сегодня разработка одного нового препарата в среднем стоит около миллиарда долларов? И если нет, то какое вообще значение, помимо фундаментального, имеет появление нового алгоритма? Если посмотреть на спектр экспертных мнений по этому вопросу, то становится ясно, что ответа на него, похоже, пока вообще никто не знает.
Про разработку лекарств
Если взять совсем общую картину, то в поиске новых лекарств можно выделить два генеральных подхода — прямой и обратный. Прямой путь представляет собой «метод проб и ошибок» — анализ огромных библиотек малых молекул на клеточных культурах или на животных для поиска нужного нам эффекта. Обратный путь, или рациональный дизайн (structure-based drug design), учитывает знания о структуре потенциальных мишеней и пытается подобрать для нужных мишеней подходящий препарат. Именно для последнего подхода потенциально пригодился бы AlphaFold2 — чтобы тестировать роботами миллионы молекул в миллионах чашек, никаких фундаментальных проблем решать не надо, бери и делай. Это долго, дорого и плохо масштабируется — а вот в рамках рационального подхода для того, чтобы проверить, взаимодействует ли лекарство с мишенью нужным способом и способно оно вызвать нужный эффект можно использовать компьютерное моделирование, для которого не нужно ничего кроме вычислительной мощности (которая дешевеет очень быстро). Кроме того, моделирование может подсказать, например, какие химические группы нужно еще навесить на лекарство, чтобы сильнее соединилось со своей мишенью. В идеале, знание структуры может дать нам информацию о лекарстве еще до того, как оно синтезировано.
Вообще, такой подход применяется в фармацевтике еще с 1990-х годов. Первым препаратом, при разработке которого учитывалась структура мишени, считается дорзоламид — лекарство от глаукомы, представляющее собой ингибитор фермента карбангидразы, которое вышло на рынок в 1994 году. За ним последовали множество других препаратов, например, блокаторы ферментов вируса иммунодефицита человека и препарат от лейкемии иматиниб, который подавляет активность тирозиновой протеинкиназы — белка, передающего стимулирующие сигналы к делению клеток. В настоящее время моделирование активно используется в разработке лекарственных препаратов. На первый взгляд, быстрое предсказание структур действительно может помочь фармацевтам. Однако проблема в том, что ускорить с помощью вычислительных методов можно лишь самый первый этап в разработке лекарств — а в реальности их, конечно, гораздо больше.
Разберемся немного подробнее, как происходит рациональный дизайн лекарств и что происходит после того, как выбрана молекула — кандидат на роль лекарства. Допустим, мы предсказали структуру нового фермента, работу которого нужно заблокировать. Мы можем взять виртуальную библиотеку разных, уже синтезированных молекул, и провести виртуальный скрининг внутри компьютера — попробовать подобрать вещество, которое бы подошло к активному центру фермента как ключ к замку. Здесь мы столкнемся с первой проблемой — нужно знать, где находится этот активный центр. Его положение тоже надо предсказать, или определить экспериментально. Кроме того, молекулы, модифицирующие активность фермента, могут связываться не в активном центре, а где-то еще — предсказать такое еще сложнее. Таким образом, даже чтобы провести первый этап анализа, нужно иметь некий экспериментальный бэкграунд вне компьютера, чтобы узнать о мишени как можно больше.
Кроме предсказания структуры мишени нам понадобятся и другие вычислительные инструменты — например, для докинга, то есть «подгонки» кандидатных молекул под нужный участок белка, расчета силы связывания молекул и молекулярного моделирования, которое поможет узнать, как белок себя ведет в растворе. Эти задачи AlphaFold2 уже не решает, и инструменты, которые тут используются, еще очень далеки от совершенства.
Когда мы решили эти проблемы и выбрали кандидатную молекулу при помощи компьютера, хорошо бы еще подтвердить взаимодействие экспериментально, в пробирке. Для этого нужно наработать в бактериях исследуемый фермент, или хотя бы тот фрагмент, с которым будет взаимодействовать лекарство, и исследовать его в комплексе с ним каким-нибудь физическим методом (например, при помощи ядерно-магнитного резонанса). Если данные подтверждают расчеты, можно приступать к исследованиям на клеточных культурах и животных. В первую очередь это нужно для того, чтобы проверить, не взаимодействует ли потенциальное лекарство с чем-то еще в клетке, и не приведет ли это взаимодействие к нежелательной токсичности. На этом этапе множество кандидатных молекул, скорее всего, отсеется.
На этапе доклинических исследований на животных нужно будет оценить то, как долго наше лекарство живет в организме, по каким органам распределяется, и конечно, какие системные эффекты оказывает, — в первую очередь, полезно ли оно при модельном заболевании. На этом этапе многие вещества тоже отсеются — может оказаться, например, что то, что должно было стать лекарством от болезни Альцгеймера и работать в нейронах головного мозга, на самом деле не проходит через гемато-энцефалический барьер, и даже не достигает своей мишени — предсказать это in silico не может ни AlphaFold2, ни какая-либо другая нейросеть.
Если же вещество успешно прошло доклинические испытания на животных, можно переходить к испытаниям на людях. Здесь мы можем ожидать дополнительные сюрпризы — токсичность, побочные эффекты, которые не были видны на животных, отсутствие терапевтического действия. По оценкам, на этапе клинических испытаний проваливается 90% препаратов, которые успешно дошли до этой стадии разработки. Получается, что проблема предсказания структуры в разработке лекарств стоит далеко не на первом месте — это значит, что, несмотря на существенный прогресс, в фармацевтике революции пока ожидать не приходится.
Однако это не означает, что программы, подобные AlphaFold2, не смогут ускорить весь описанный цикл. Как выразился специалист по разработке препаратов Дерек Лоув (Derek Lowe), комментируя успех Google в блоге на страницах журнала Science Translational Medicine, главное, что он хотел бы получить от компьютерных методов — «это быстрее и дешевле понимать, что мы в очередной раз выбрали не ту мишень для лекарства». По его мнению, основная проблема в поиске потенциальных препаратов — не определение структур белков, а недостаточное количество знаний о механизмах болезней, где они задействованы. Нужно детально представлять себе, как организм работает в норме и при патологии и только тогда можно будет найти правильные мишени для лекарств. Но здесь даже самыми сложными нейросетями не обойтись — нужны ученые, люди, которые каждый день с утра до вечера исследуют работу живых организмов — такие, как сам Дерек Лоув, Мария Соколова и их коллеги. А потому главное значение, которые будет иметь появление AlphaFold2, будет заключаться в том, что теперь программа сможет хоть немного упростить жизнь этих людей.
Отдел «Разбор»